| |
|
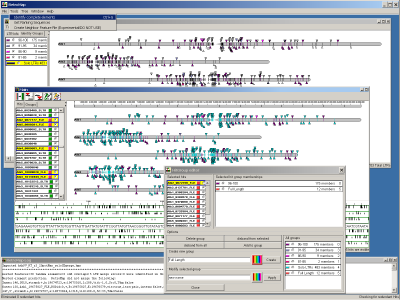
Introduction
RetroMap is an application designed to help characterize LTR retroelements on a genome scale in a visually
interactive manner. This is NOT a particularly great for comprehensive identification of elements found in highly nested
contexts. Only the most internal element of those which are nested is likely to be identified as a complete element. The other
elements will be treated as solo LTRs and internal regions depending on how you have set up your element searches. However, it is
still a handy way to get a quick visual overview of the situation and create pretty figures.
Any significant future changes will be reflected in this document. Please note that this software is highly experimental
and has not been published yet though it is mentioned briefly in my 2004 Genome Biology paper. Until noted otherwise in
this document, any citations should refer to the Genome Biology manuscript. I will work to address any problems you
may have and incorporate your suggestions and requests for features as best as I am able.
-=Brooke P-B
System Requirements
RetroMap is written in the Java language for enhanced cross-platform compatability. However, there are currently a few
external dependencies which have to be met.
- First, ensure that you are using the most recent version of the RetroMap program which may be downloaded from
www.burchsite.com/bioi/java/RetroMap.jar. The current
version at this time is 0.021. You may check the version you have by selecting
'Help->About' which will display a simple dialog showing the program version. This will ensure that you have the
latest features and bug fixes. Second, it is necessary that the machine running RetroMap knows how to run Java
programs. If it does not run at all upon starting the executable RetroMap.jar file, then it is likely you will need
to install a copy of the free Java
Runtime from SUN. This MUST be version 5 or higher (may be listed as version 1.5)
- The free
NCBI BLAST StandAlone software or at least the Blast2Sequences program of the BLAST suite must be installed in
order to use the automatic LTR identification feature. If you are going to follow the tutorial below, then you will
need to install the rest of the BLAST executables anyhow. In the future, I intend to add internal search functions
to eliminate this reliance on external software.
- RAM and lots of it when working with large datasets. The more the better. Hardware upgrades are beyond the scope
of this document
Tutorial
Concepts
RetroMap is set up to allow datasets to be combined with each other as the default. It is not very good at reversing the
process so I would suggest that you keep that in mind when choosing when and what to save. This also means that
the software will quite happily append data to existing files without warning you under the belief that it is your intention to
do so.
Hidden data: some objects in the window allow you to hide them. RetroMap considers these objects (while hidden) to be
completely unavailable to it. This means that information for hidden objects will not be saved to files or included
in any search functions you perform while hidden. This is the only mechanism available for removing information from sessions
and saved files.
Importing data files
RetroMap accepts several different file formats as input. It will automatically try to determine which file type you are
attempting to import. If the file is not supported by this program the import will fail.
BLAST XML formatted output files are the primary source for data. RetroMap will import all BLAST hits from the XML file and
if necessary, condense overlapping hits into a single coverage format where overlapping hits are combined into a new hit with
boundaries encompassing the greatest possible extent for the two hits.
The native RetroMap format is also XML based and consists of two types. After a BLAST file has been imported your work may
be saved as a RetroMap (*.hmx) file. A specialized (*.hgd) XML format file is allowable to impose genome information such as
centromere locations upon your dataset so they may be drawn. This file also typically contains information about sequences
used to construct the BLAST database which was queried. Genome data files, pre-formatted BLAST databases , and source
chromosome sequences are available for Arabidopsis thaliana, Drosophila melanogaster,
Saccharomyces cerevisiae, and Schizosaccharomyces pombe
Coordinates provided by RetroMap are relative to the orientation that the reference sequences are found in. An object
found on the antisense strand relative to the reference sequence will have a start coordinate which is larger than the end
coordinate.
Phylogenetic information may be applied to the hits through import of a MEGA tree (.tre NOT a tree session file.mts). Working
with MEGA tree files will be covered in the Phylogentic data section when I create it.
A step by step example
Here I'll walk you through everything you need to do to work with the features of the program using example data files which may be
downloaded as indicated below.
- Prepare the sequences for the blast search. *NOTE* If you are just wanting to get a quick overview, the first 2 steps can be replaced
with an XML BLAST search result file generated using the NCBI BLAST search pages for example. Those who are looking for a more
interactive experience should follow along with this Drosophila chromosome arm example. You may use your own sequences in place
of the Drosophila ones discussed here.
- Download the Drosophila melanogaster sequences.
- Rename the fasta headers for each sequence to something short but unique. This name is what RetroMap will use as the label
for the large chromosome sequences or contigs. examples of good identifiers are things like Chr1, or Dm_2L. The fasta
header lines in each file should now look something like '>Dm_2L'
- Create a large text document where you have appended all of the fasta sequences together. The file created for this
example was called DmelGenomeV4.mfa. Tip: If you would like sequence retrieval to be as speedy as possible, ensure that the fasta
files are set up to be as minimal as possible. This means that the header line should be followed by the sequence line where the
sequence is all on a single line and contains no non-dna characters.
- Generate a BLAST database for the genome sequences. Blast commands will have to be run from a command or terminal prompt, see the
Blast documentation for further details on available commands and settings. formatdb was used with the following command line:
'formatdb -t "D. melanogaster genome rel. 4 blastdb created 2004-11-04" -i "DmelGenomeV4.mfa" -p F -o T -n DmelGenomeV4'
- Get an internal (located between the LTRs) known sequence
to use as a blast query sequence to identify new retrotransposons. Save it as Endovir_IN.fan. This is a core (conserved region)
of the integrase gene from the Arabidopsis thaliana Endovir1-1 Pseudoviridae retrotransposon.
- Run a first round blast search making sure you have set blast to generate XML output for the report. My command was:
'blastall -p tblastn -d DmelGenomeV4 -i Endovir_IN.fan -m 7-e 1e-5 -o IN_Rnd1.xml -F F -v 0 -b 1000000'
Alternatively, a fasta file containing multiple fasta formatted sequences can be used for querying the database with a number of query sequences
at the same time.
- Begin a RetroMap session
- Start RetroMap by double clicking on it for Windows and Macs or typing 'java -jar RetroMap.jar' in the directory where
RetroMap.jar is located for Unix command lines. If either of these do not work, please contact your system administrator or computer
geek friend to ensure that java is properly installed and the java.exe executable can be found in your system path.
- Select File->Import (Ctrl+I) to open a file dialog. Navigate to the blast report you wish to use and click on Open.
Note that this blast report file MUST be in XML format which you should have selected when running BLAST with the '-m 7' argument.
- A rudimentary 'Blast Import Options' window should open. For now just look at the 'Default output filename' and change it if you like with
the 'Select Filename' button. This is the root save location and name for all RetroMap generated files. Click on 'OK' to start the import
process.
- Create a large text document where you have appended all of the fasta sequences together. The file created for this
example was called DmelGenomeV4.mfa.
- If all went well with the import, a new window called 'Main' should have opened on the RetroMap desktop displaying the locations of
all blast hits matching a reference sequence (subject in blast parlance and is the chromosome sequence in this example) from the database
on their respective reference sequence. You will probably want to adjust the size and position of this window.
- Problems with the import may be indicated by symptoms such as all hits being displayed on only one strand of the query sequence(s) and/or
'No definition line found' may be displayed on items throughout the window. This usually means that the importer did not find the XML
document to be structured the way it knew how to handle. If these problems recurr, then you may want to contact me to fix this as I consider
it to be a bug. Please include a copy of the problematic XML file with your report.
- If you wish, you may save the current HitPlot (minus the scalebar currently, :-( sorry) by right clicking (or whatever needs to be done on your operating
system to display popup menus) and click on 'Save Image'. Doing so will save a scalable vector graphic (.svg) file in
the current directory with the same name as that you selected during the import with a '.svg' extension added. This may
be opened and worked with by image editors capable of viewing SVG files such as Adobe Illustrator&tm;.
- Go ahead and save the current data set so that you can revert to it if necessary. To save, follow these steps:
- Select File->Save or Ctrl+S to open the save dialog
- Select the 'Save HitMapper (hmx) data' and 'Save seq for hits' option buttons. You will see that the output names have
now been set for those options. If you would like to change the base name for the output files you can do so by
clicking on the 'Change File' button. RetroMap native data will be saved to files with a '.hmx' extension while the
sequences will be '.fan'
- When you are satisfied with the Save options, click on the 'Continue' button. Since RetroMap doesn't know where the
source sequence files are located, it will prompt you to provide the source file for each sequence. The sequences may
be contained in a single multi-FASTA file or in individual files. The file chooser title lists the name of the sequence
that it currently wishes you provide. The sequence MUST be in FASTA format and have a header beginning with the sequence
name, e.g. '>SeqName Possibly other FASTA header info'. In this example, the title of my chooser says it is looking for
Dm_2L. Navigate to the source file, select it and click the 'Open' button. If you are following along with my example,
the source file will be named DmelGenomeV4.mfa. If you aren't, then you may have to provide the filenames for a number of
sequences.
- It may take a while for RetroMap to index the fasta file (particularly for genome sized ones like the Drosophila example file)
and the application will be unresponsive until indexing completes. The status bar at the bottom of the application will
indicate when indexing has completed and tell you the number of sequences written. As long as the file and it's location
do not change, the indexing should only have to occur once. Please note that if a sequence file already exists, RetroMap
will append to the end of it rather than replace it. However, the (.hmx) file WILL be replaced.
- Now we can attempt to find LTRs for all of the imported sequences. Select Tools->'Identify Complete Elements' or Ctrl+G to
open the full-length element (LTR to LTR) identification dialog.
- If you are following along with the example, the dialog will appear and show a list of the reference sequences along with
their file locations. If not, the dialog will ask that these be set using the 'Select file' button to tell RetroMap where
the requested (if the blast file provides a name) source sequence file for the hits is located on your computer.
- Set a default name for the output file, eg. test. The extensions are added automatically and noted in the dialog. If you
fail to provide one, the default filename will be set to 'default', originally enough.
- Select the 'Save full length elements?' radio button and any others you'd like. This is currently the only chance you have
to save the sequences for these.
- Click Continue. Since you haven't told the program where to find NCBI's bl2seq program yet, it will prompt you to find the
file. Navigate to it and hit 'Open'. RetroMap will now start searching for LTRs for each of the blast hits you
imported. Therefore, you really, really wouldn't like to use LTRs as the queries for creating the blast report you
import.
- RetroMap will appear to freeze while it performs the search. The text in the lower left of the status bar will say
"Completed LTR Search" when the program finishes looking for LTRs. At this point you can continue to work with the
program.
- Full-length sequences for those hits which appear to be part of elements with two LTRs will be found in the output files you
selected.
- Selecting the 'Save full length elements' button is going to
make RetroMap take a long time to retrieve the sequences, several minutes on my 2GHz 64bit machine. This is because
RetroMap assumes that your sequence file (DmelGenomeV4.mfa) is potentially full of non-sequence characters like spaces,
newlines, and numbers. RetroMap is currently set up to minimize the amount of disk space it consumes when running so it
doesn't reformat your input sequence file into a version which enables much more efficient sequence retrieval. This would
entail creating new copies of your sequence files which could consume a lot of space if you were working with a large
eukaryotic genome. In the future I may add an option (or requirement!) to have RetroMap ensure that sequence files are
formatted the way enabling fast sequence retrieval.
- RetroMap can output its information on the objects it is displaying as a tab-delimited output file (*.tdf). This file is suitable
for import into spreadsheet software such as MS Excel or Gnumeric
- To do this select 'File->Save...'. This re-opens the save dialog we used above. This time select the
'Tab delimited data file' option. Change the filename if you need to and hit 'Continue'.
- Open the new file with a spreadsheet application. I'm writing this tutorial on a linux machine right now, so I am going to
use Gnumeric. You may have to go through an import dialog in your spreadsheet application to tell it that the data is in
tab-delimited format.
- The top row is a header line providing headings for each of the columns in the table. The definitions follow.
| HitName | The name that has been assigned to a particular hit object |
| RefSeq | This will be the sequence name that this object belongs to and is located on |
| Strand |
A (+) or (-) symbol representing the orientation of the hit object relative to the RefSeq as
it is found in the source fasta file. |
| HitBeg | The position on the the RefSeq where this hit object begins in it's own sense oriention |
| HitEnd |
The position on the the RefSeq where this hit object begins in it's own sense oriention. This means that a hit
on the RefSeqs antisense strand will have a begin coordinate that is larger than its end coordinate. |
| HitLength |
The length of the original hit. |
| LTR | This can be 5', 3', Solo, or unset |
| LTRbeg |
Provides the begin coordinate on the RefSeq if this row provides data about an LTR. Empty otherwise. |
| LTRend | The end coordinate for this LTR if it is an LTR row |
| Tandem | True or False. True indicates that RetroMap believes that two elements share a LTR |
| Internal |
True or False. True if this hit object is entirely contained or nested in another hit object's
sequence |
| LTRscore |
Higher scores are better. This represents how likely RetroMap believes the selected LTR is to be a genuine
LTR. Currently it only: 1) checks to see if the LTR sequences begin with TG or TA and end with TCA or CA. 2) checks
whether these residues are identical on both LTRs. Future improvements would include searching for a target-site
duplication, and nearby primer binding sites |
| LTRlength |
Actual residue length of this particular LTR. Non-identical LTRs may have different lengths |
| numID | The number of identical aligned residues for the two LTRs |
| totalCompared | The length of the alignment between the two LTRs |
| %ID | ((numID / totalCompared) * 100) |
| AgeEst |
If you have provided a nucleotide substitution rate, RetroMap provides an age estimate in MYr for the time
since an element with non-identical LTRs inserted. |
| ElementLength | This encompasses the largest known bounds for the hit which means that for a hit with
ltrs, it will represent the span from the beginning of the 5'LTR to the end of the 3'LTR |
| grp:groupname |
This column will note members of a group you have set up by listing the group name next to members of that
group. There will be as many of these columns as there are groups. |
Bugs and Desired Enhancements
Bugs
Enhancements
- Web interactivity for reference sequence imports
- Custom chromosomal and element tag construction
- Turn off hit merging across queries so that hits remain distinct by query
- Move the save options from the Find complete elements dialog to the save menu
|
|
|